flip and flip_seqs reverse-complement specified bins or individual
sequences and their features. sync automatically flips bins using a
heuristic that maximizes the amount of forward strand links between
neighboring bins.
Usage
flip(x, ..., .bin_track = seqs)
flip_seqs(x, ..., .bins = everything(), .seq_track = seqs, .bin_track = seqs)
sync(x, link_track = 1, min_support = 0)Arguments
- x
a gggenomes object
- ...
bins or sequences to flip in dplyr::select like syntax (numeric position or unquoted expressions)
- .bin_track, .seq_track
when using a function as selector such as
tidyselect::where(), this specifies the track in which context the function is evaluated.- .bins
preselection of bins with sequences to flip. Useful if selecting by numeric position. It sets the context for selection, for example the 11th sequences of the total set might more easily described as the 2nd sequences of the 3rd bin:
flip_seqs(2, .bins=3).- link_track
the link track to use for flipping bins nicely
- min_support
only flip a bin if at least this many more nucleotides support an inversion over the given orientation
Details
For more details see the help vignette:
vignette("flip", package = "gggenomes")
Examples
library(patchwork)
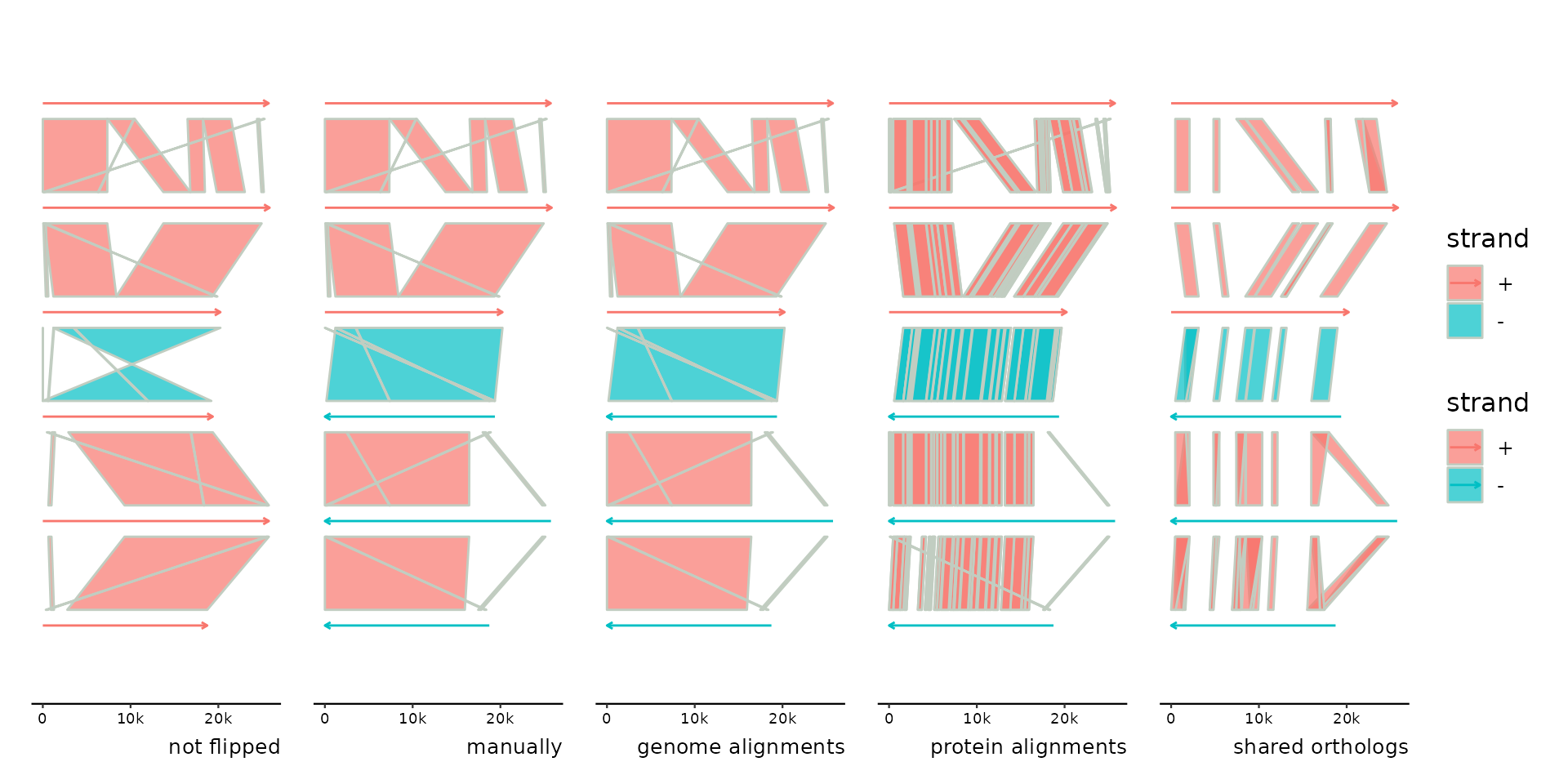
p <- gggenomes(genes = emale_genes) +
geom_seq(aes(color = strand), arrow = TRUE) +
geom_link(aes(fill = strand)) +
expand_limits(color = c("-")) +
labs(caption = "not flipped")
#> No seqs provided, inferring seqs from feats
# nothing flipped
p0 <- p %>% add_links(emale_ava)
# flip manually
p1 <- p %>%
add_links(emale_ava) %>%
flip(4:6) + labs(caption = "manually")
# flip automatically based on genome-genome links
p2 <- p %>%
add_links(emale_ava) %>%
sync() + labs(caption = "genome alignments")
#> Flipping: E4-10_086,E4-10_112,RCC970_016B
# flip automatically based on protein-protein links
p3 <- p %>%
add_sublinks(emale_prot_ava) %>%
sync() + labs(caption = "protein alignments")
#> Transforming sublinks with "aa2nuc". Disable with `.transform = "none"`
#> Flipping: E4-10_086,E4-10_112,RCC970_016B
# flip automatically based on genes linked implicitly by belonging
# to the same clusters of orthologs (or any grouping of your choice)
p4 <- p %>%
add_clusters(emale_cogs) %>%
sync() + labs(caption = "shared orthologs")
#> Joining with `by = join_by(feat_id)`
#> Flipping: E4-10_086,E4-10_112,RCC970_016B
p0 + p1 + p2 + p3 + p4 + plot_layout(nrow = 1, guides = "collect")