## Loading required package: ggplot2
## gggenomes v1.1.3
##
## If you use 'gggenomes' in published research, please cite:
##
## Hackl T, Ankenbrand M, van Adrichem B, Wilkins D, Haslinger K (2024).
## "gggenomes: effective and versatile visualizations for comparative
## genomics." _arXiv_. doi:10.48550/arXiv.2411.13556
## <https://doi.org/10.48550/arXiv.2411.13556>.
##
## Attaching package: 'gggenomes'
## The following object is masked from 'package:graphics':
##
## layout
## No seqs provided, inferring seqs from feats
## Flipping: E4-10_086,E4-10_112,RCC970_016B
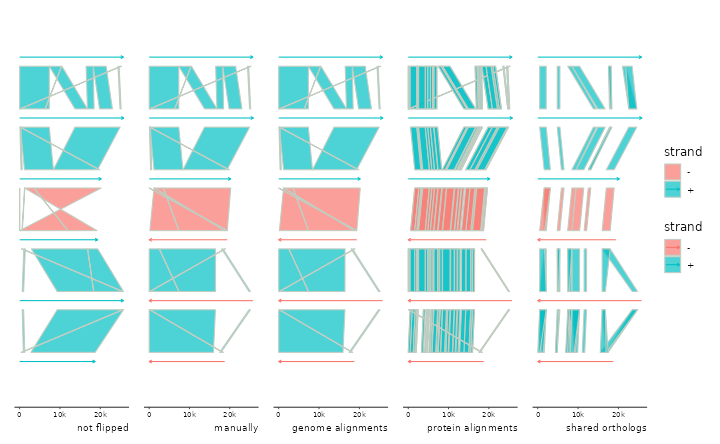
# flip automatically based on protein-protein links
p3 <- p %>% add_sublinks(emale_prot_ava) %>%
sync() + labs(caption="protein alignments")
## Transforming sublinks with "aa2nuc". Disable with `.transform = "none"`
## Flipping: E4-10_086,E4-10_112,RCC970_016B
# flip automatically based on genes linked implicitly by belonging
# to the same clusters of orthologs (or any grouping of your choice)
p4 <- p %>% add_clusters(emale_cogs) %>%
sync() + labs(caption="shared orthologs")
## Joining with `by = join_by(feat_id)`
## Flipping: E4-10_086,E4-10_112,RCC970_016B
p0 + p1 + p2 + p3 + p4 + plot_layout(nrow=1, guides="collect")
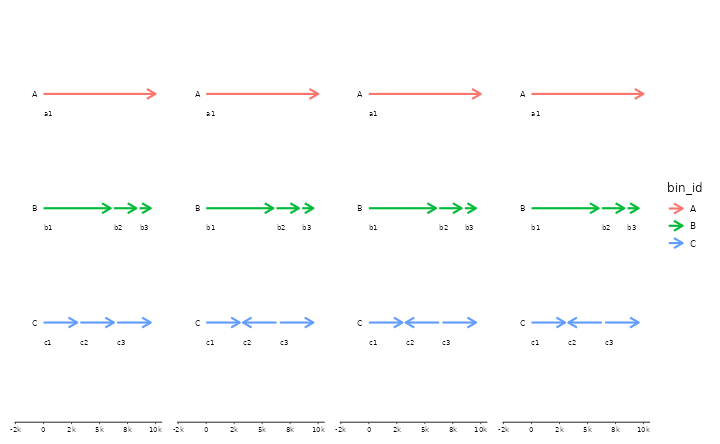
# flip seqs inside bins
s0 <- tibble::tibble(
bin_id = c("A", "B", "B", "B", "C", "C", "C"),
seq_id = c("a1","b1","b2","b3","c1","c2","c3"),
length = c(1e4, 6e3, 2e3, 1e3, 3e3, 3e3, 3e3))
p <- gggenomes(seqs=s0) +
geom_seq(aes(color=bin_id), size=1, arrow = arrow(angle = 30, length = unit(10, "pt"),
ends = "last", type = "open")) +
geom_bin_label() + geom_seq_label() +
expand_limits(color=c("A","B","C"))
## Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
## ℹ Please use `linewidth` instead.
## ℹ The deprecated feature was likely used in the gggenomes package.
## Please report the issue at <https://github.com/thackl/gggenomes/issues>.
## This warning is displayed once per session.
## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
## generated.
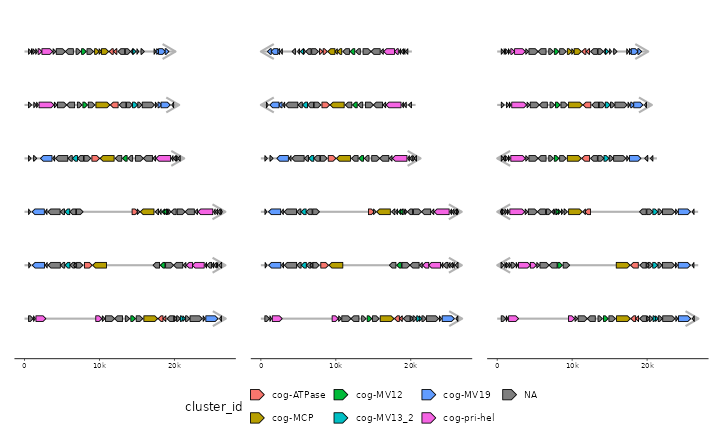
# fancy flipping using tidyselect::where for dynamic selection
p <- gggenomes(emale_genes,emale_seqs) %>% add_clusters(emale_cogs) +
geom_seq(color="grey70", linewidth=1, arrow = arrow(angle = 30, length = unit(15, "pt"),
ends = "last", type = "open")) +
geom_gene(aes(fill=cluster_id))
## Joining with `by = join_by(feat_id)`
# flip all short seqs - where() applied to .bin_track=seqs
p1 <- p %>% flip(where(~.x$length < 21000))
# flip all seqs with MCP on "-" - where() applied to .bin_track=genes
p2 <- p %>% flip(where(~any(.x$strand[.x$cluster_id %in% "cog-MCP"] == "-")), .bin_track=genes)
p + p1 + p2 + plot_layout(nrow=1, guides="collect") & theme(legend.position = "bottom")