Put bin labels left of the sequences. nudge_left adds space relative to the
total bin width between the label and the seqs, by default 5%. expand_left
expands the plot to the left by 20% to make labels visible.
Usage
geom_bin_label(
mapping = NULL,
data = bins(),
hjust = 1,
size = 3,
nudge_left = 0.05,
expand_left = 0.2,
expand_x = NULL,
expand_aes = NULL,
yjust = 0,
...
)Arguments
- mapping
Set of aesthetic mappings created by
aes(). If specified andinherit.aes = TRUE(the default), it is combined with the default mapping at the top level of the plot. You must supplymappingif there is no plot mapping.- data
The data to be displayed in this layer. There are three options:
If
NULL, the default, the data is inherited from the plot data as specified in the call toggplot().A
data.frame, or other object, will override the plot data. All objects will be fortified to produce a data frame. Seefortify()for which variables will be created.A
functionwill be called with a single argument, the plot data. The return value must be adata.frame, and will be used as the layer data. Afunctioncan be created from aformula(e.g.~ head(.x, 10)).- hjust
Moves the text horizontally
- size
of the label
- nudge_left
by this much relative to the widest bin
- expand_left
by this much relative to the widest bin
- expand_x
expand the plot to include this absolute x value
- expand_aes
provide custom aes mappings for the expansion (advanced)
- yjust
for multiline bins set to 0.5 to center labels on bins, and 1 to align labels to the bottom.
- ...
Other arguments passed on to
layer()'sparamsargument. These arguments broadly fall into one of 4 categories below. Notably, further arguments to thepositionargument, or aesthetics that are required can not be passed through.... Unknown arguments that are not part of the 4 categories below are ignored.Static aesthetics that are not mapped to a scale, but are at a fixed value and apply to the layer as a whole. For example,
colour = "red"orlinewidth = 3. The geom's documentation has an Aesthetics section that lists the available options. The 'required' aesthetics cannot be passed on to theparams. Please note that while passing unmapped aesthetics as vectors is technically possible, the order and required length is not guaranteed to be parallel to the input data.When constructing a layer using a
stat_*()function, the...argument can be used to pass on parameters to thegeompart of the layer. An example of this isstat_density(geom = "area", outline.type = "both"). The geom's documentation lists which parameters it can accept.Inversely, when constructing a layer using a
geom_*()function, the...argument can be used to pass on parameters to thestatpart of the layer. An example of this isgeom_area(stat = "density", adjust = 0.5). The stat's documentation lists which parameters it can accept.The
key_glyphargument oflayer()may also be passed on through.... This can be one of the functions described as key glyphs, to change the display of the layer in the legend.
Examples
s0 <- read_seqs(list.files(ex("cafeteria"), "Cr.*\\.fa.fai$", full.names = TRUE))
#> Reading 'fai' with `read_fai()`:
#> * file_id: CrBVI.fa [/home/runner/work/_temp/Library/gggenomes/extdata/cafeteria/CrBVI.fa.fai]
#> * file_id: CrCflag.fa [/home/runner/work/_temp/Library/gggenomes/extdata/cafeteria/CrCflag.fa.fai]
#> * file_id: CrE410P.fa [/home/runner/work/_temp/Library/gggenomes/extdata/cafeteria/CrE410P.fa.fai]
#> * file_id: CrRCC970.fa [/home/runner/work/_temp/Library/gggenomes/extdata/cafeteria/CrRCC970.fa.fai]
s1 <- s0 %>% dplyr::filter(length > 5e5)
gggenomes(emale_genes) + geom_seq() + geom_gene() +
geom_bin_label()
#> No seqs provided, inferring seqs from feats
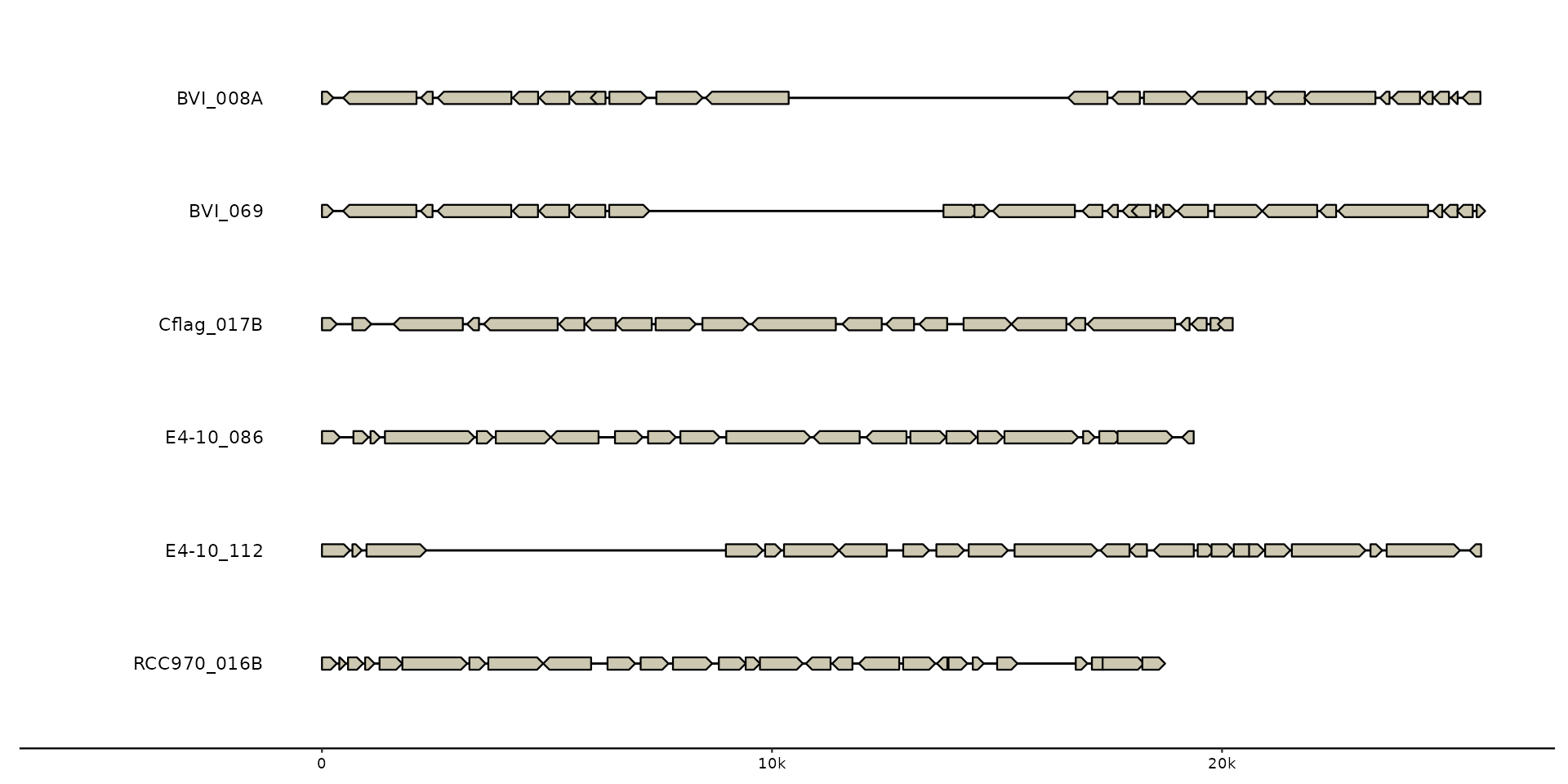 # make larger labels and extra room on the canvas
gggenomes(emale_genes) + geom_seq() + geom_gene() +
geom_bin_label(size = 7, expand_left = .4)
#> No seqs provided, inferring seqs from feats
# make larger labels and extra room on the canvas
gggenomes(emale_genes) + geom_seq() + geom_gene() +
geom_bin_label(size = 7, expand_left = .4)
#> No seqs provided, inferring seqs from feats
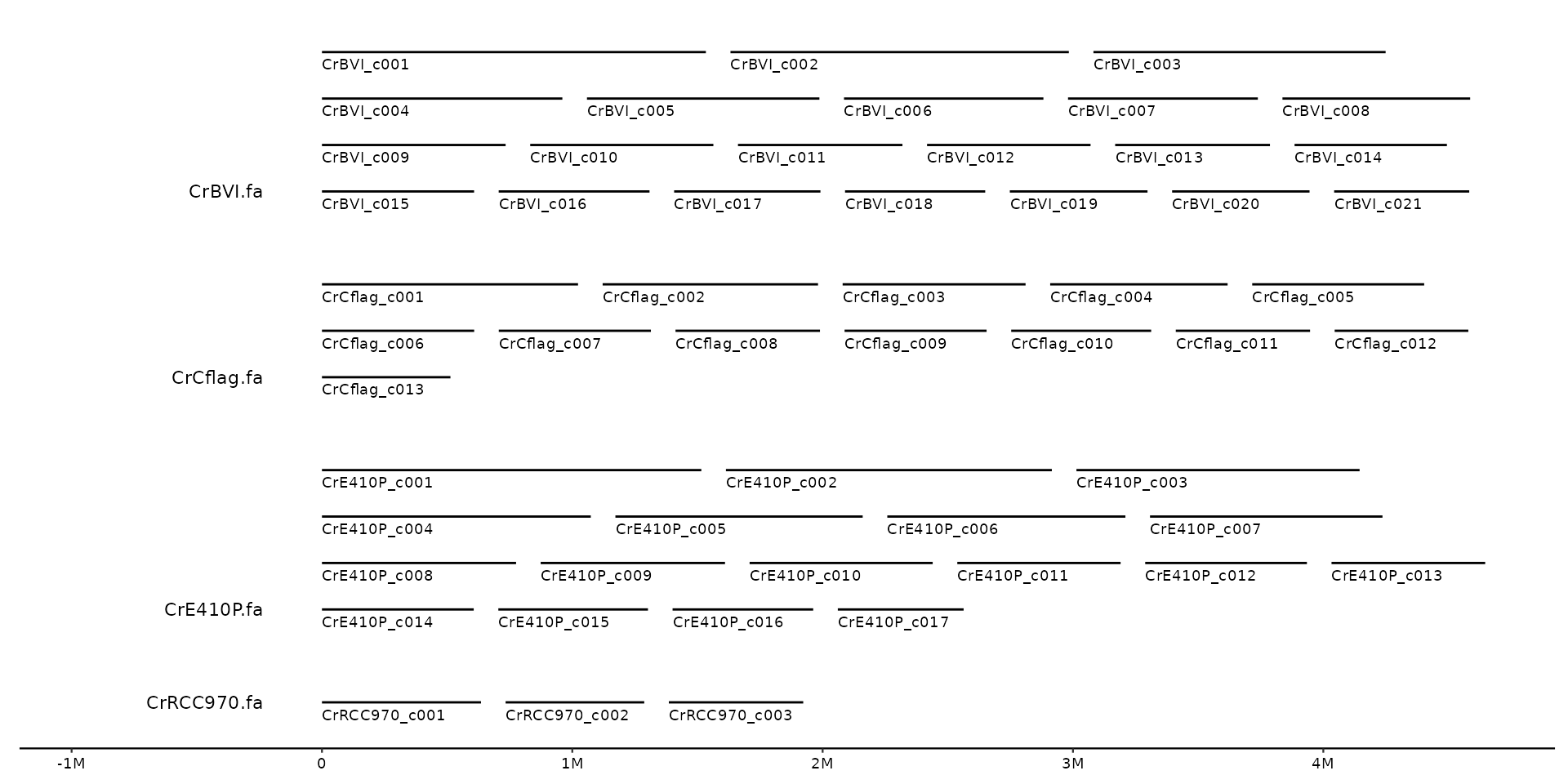
 # align labels for wrapped bins:
# top
gggenomes(seqs = s1, infer_bin_id = file_id, wrap = 5e6) +
geom_seq() + geom_bin_label() + geom_seq_label()
# align labels for wrapped bins:
# top
gggenomes(seqs = s1, infer_bin_id = file_id, wrap = 5e6) +
geom_seq() + geom_bin_label() + geom_seq_label()
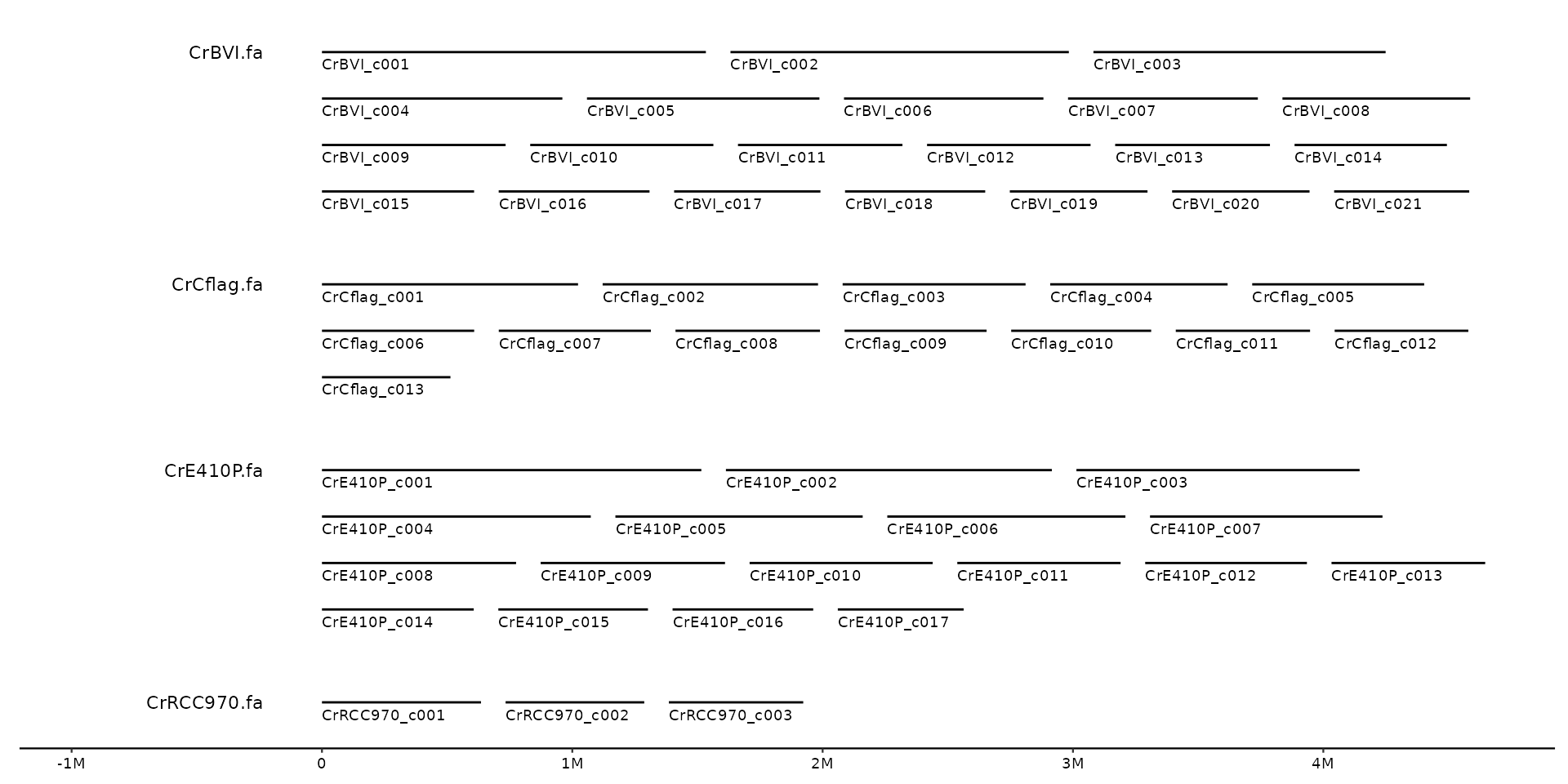 # center
gggenomes(seqs = s1, infer_bin_id = file_id, wrap = 5e6) +
geom_seq() + geom_bin_label(yjust = .5) + geom_seq_label()
# center
gggenomes(seqs = s1, infer_bin_id = file_id, wrap = 5e6) +
geom_seq() + geom_bin_label(yjust = .5) + geom_seq_label()
 # bottom
gggenomes(seqs = s1, infer_bin_id = file_id, wrap = 5e6) +
geom_seq() + geom_bin_label(yjust = 1) + geom_seq_label()
# bottom
gggenomes(seqs = s1, infer_bin_id = file_id, wrap = 5e6) +
geom_seq() + geom_bin_label(yjust = 1) + geom_seq_label()