Track selection works like dplyr::pull() and supports unquoted ids and
positional arguments. ... can be used to subset the data in
dplyr::filter() fashion. pull-prefixed variants return the specified
track from a gggenomes object. Unprefixed variants work inside geom_* calls.
Usage
feats(.track_id = 1, ..., .ignore = "genes", .geneify = FALSE)
feats0(.track_id = 1, ..., .ignore = NA, .geneify = FALSE)
genes(..., .gene_types = c("CDS", "mRNA", "tRNA", "tmRNA", "ncRNA", "rRNA"))
links(.track_id = 1, ..., .ignore = NULL, .adjacent_only = NULL)
seqs(...)
bins(..., .group = vars())
track(.track_id = 1, ..., .track_type = NULL, .ignore = NULL)
pull_feats(.x, .track_id = 1, ..., .ignore = "genes", .geneify = FALSE)
pull_genes(
.x,
...,
.gene_types = c("CDS", "mRNA", "tRNA", "tmRNA", "ncRNA", "rRNA")
)
pull_links(.x, .track_id = 1, ..., .ignore = NULL, .adjacent_only = NULL)
pull_seqs(.x, ...)
pull_bins(.x, ..., .group = vars())
# S3 method for class 'gggenomes_layout'
pull_bins(.x, ..., .group = vars())
pull_track(.x, .track_id = 1, ..., .track_type = NULL, .ignore = NULL)Arguments
- .track_id
The track to pull out, either as a literal variable name or as a positive/negative integer giving the position from the left/right.
- ...
Logical predicates passed on to dplyr::filter. "seqs", "feats", "links". Affects position-based selection.
- .ignore
track names to ignore when selecting by position. Default is "genes", if using
feats0this defaults toNA.- .geneify
add dummy type, introns and geom_id column to play nicely with geoms supporting multi-level and spliced gene models.
- .gene_types
return only feats of this type (
type %in% .gene_types)- .adjacent_only
filter for links connecting direct neighbors (
abs(y-yend)==1))- .group
what variables to use in grouping of bins from seqs in addition to
yandbin_id. Use this to get additional shared variables from the seqs table into the bins table.- .track_type
restrict to these types of tracks - any combination of "seqs", "feats", "links".
- .x
A gggenomes or gggenomes_layout object.
Value
A function that pulls the specified track from a gggenomes object.
A function that pulls the specified track from a gggenomes object.
A function that pulls the specified track from a gggenomes object.
A function that pulls the specified track from a gggenomes object.
A function that pulls the specified track from a gggenomes object.
A function that pulls the specified track from a gggenomes object.
A function that pulls the specified track from a gggenomes object.
Functions
feats(): by default pulls out the first feat track not named "genes".feats0(): by default pulls out the first feat track.genes(): pulls out the first feat track (genes), filtering for records withtype=="CDS", and adding a dummygene_idcolumn if missing to play nice with multi-exongeoms.links(): by default pulls out the first link track.seqs(): pulls out the seqs track (there is only one).bins(): pulls out a binwise summary table of the seqs data poweringgeom_bin_*()calls. The bin table is not a real track, but recomputed on-the-fly.track(): pulls from all tracks in order seqs, feats, links.
Examples
gg <- gggenomes(emale_genes, emale_seqs, emale_tirs, emale_ava)
gg %>% track_info() # info about track ids, positions and types
#> # A tibble: 4 × 4
#> # Groups: type [3]
#> id type i n
#> <chr> <chr> <int> <dbl>
#> 1 seqs seqs 1 6
#> 2 genes feats 1 143
#> 3 feats feats 2 12
#> 4 links links 1 38
# get first feat track that isn't "genes" (all equivalent)
gg %>% pull_feats() # easiest
#> # A tibble: 12 × 20
#> y x xend bin_id seq_id start end file_id strand type feat_id
#> <int> <dbl> <dbl> <chr> <chr> <int> <int> <chr> <chr> <chr> <chr>
#> 1 2 0 473 BVI_008A BVI_0… 1 473 emales… + repe… BVI_00…
#> 2 2 26820 26347 BVI_008A BVI_0… 26348 26820 emales… - repe… BVI_00…
#> 3 3 0 488 BVI_069 BVI_0… 1 488 emales… + repe… BVI_06…
#> 4 3 26808 26320 BVI_069 BVI_0… 26321 26808 emales… - repe… BVI_06…
#> 5 4 0 1081 Cflag_017B Cflag… 1 1081 emales… + repe… Cflag_…
#> 6 4 21311 20230 Cflag_017B Cflag… 20231 21311 emales… - repe… Cflag_…
#> 7 5 0 319 E4-10_086 E4-10… 1 319 emales… + repe… E4-10_…
#> 8 5 20642 20323 E4-10_086 E4-10… 20324 20642 emales… - repe… E4-10_…
#> 9 1 0 458 E4-10_112 E4-10… 1 458 emales… + repe… E4-10_…
#> 10 1 26856 26398 E4-10_112 E4-10… 26399 26856 emales… - repe… E4-10_…
#> 11 6 0 454 RCC970_016B RCC97… 1 454 emales… + repe… RCC970…
#> 12 6 20152 19698 RCC970_016B RCC97… 19699 20152 emales… - repe… RCC970…
#> # ℹ 9 more variables: introns <list>, parent_ids <list>, source <chr>,
#> # score <chr>, phase <int>, name <chr>, width <chr>, geom_id <chr>,
#> # .marginal <lgl>
gg %>% pull_feats(feats) # by id
#> # A tibble: 12 × 20
#> y x xend bin_id seq_id start end file_id strand type feat_id
#> <int> <dbl> <dbl> <chr> <chr> <int> <int> <chr> <chr> <chr> <chr>
#> 1 2 0 473 BVI_008A BVI_0… 1 473 emales… + repe… BVI_00…
#> 2 2 26820 26347 BVI_008A BVI_0… 26348 26820 emales… - repe… BVI_00…
#> 3 3 0 488 BVI_069 BVI_0… 1 488 emales… + repe… BVI_06…
#> 4 3 26808 26320 BVI_069 BVI_0… 26321 26808 emales… - repe… BVI_06…
#> 5 4 0 1081 Cflag_017B Cflag… 1 1081 emales… + repe… Cflag_…
#> 6 4 21311 20230 Cflag_017B Cflag… 20231 21311 emales… - repe… Cflag_…
#> 7 5 0 319 E4-10_086 E4-10… 1 319 emales… + repe… E4-10_…
#> 8 5 20642 20323 E4-10_086 E4-10… 20324 20642 emales… - repe… E4-10_…
#> 9 1 0 458 E4-10_112 E4-10… 1 458 emales… + repe… E4-10_…
#> 10 1 26856 26398 E4-10_112 E4-10… 26399 26856 emales… - repe… E4-10_…
#> 11 6 0 454 RCC970_016B RCC97… 1 454 emales… + repe… RCC970…
#> 12 6 20152 19698 RCC970_016B RCC97… 19699 20152 emales… - repe… RCC970…
#> # ℹ 9 more variables: introns <list>, parent_ids <list>, source <chr>,
#> # score <chr>, phase <int>, name <chr>, width <chr>, geom_id <chr>,
#> # .marginal <lgl>
gg %>% pull_feats(1) # by position
#> # A tibble: 12 × 20
#> y x xend bin_id seq_id start end file_id strand type feat_id
#> <int> <dbl> <dbl> <chr> <chr> <int> <int> <chr> <chr> <chr> <chr>
#> 1 2 0 473 BVI_008A BVI_0… 1 473 emales… + repe… BVI_00…
#> 2 2 26820 26347 BVI_008A BVI_0… 26348 26820 emales… - repe… BVI_00…
#> 3 3 0 488 BVI_069 BVI_0… 1 488 emales… + repe… BVI_06…
#> 4 3 26808 26320 BVI_069 BVI_0… 26321 26808 emales… - repe… BVI_06…
#> 5 4 0 1081 Cflag_017B Cflag… 1 1081 emales… + repe… Cflag_…
#> 6 4 21311 20230 Cflag_017B Cflag… 20231 21311 emales… - repe… Cflag_…
#> 7 5 0 319 E4-10_086 E4-10… 1 319 emales… + repe… E4-10_…
#> 8 5 20642 20323 E4-10_086 E4-10… 20324 20642 emales… - repe… E4-10_…
#> 9 1 0 458 E4-10_112 E4-10… 1 458 emales… + repe… E4-10_…
#> 10 1 26856 26398 E4-10_112 E4-10… 26399 26856 emales… - repe… E4-10_…
#> 11 6 0 454 RCC970_016B RCC97… 1 454 emales… + repe… RCC970…
#> 12 6 20152 19698 RCC970_016B RCC97… 19699 20152 emales… - repe… RCC970…
#> # ℹ 9 more variables: introns <list>, parent_ids <list>, source <chr>,
#> # score <chr>, phase <int>, name <chr>, width <chr>, geom_id <chr>,
#> # .marginal <lgl>
gg %>% pull_feats(2, .ignore = NULL) # default .ignore="genes"
#> # A tibble: 12 × 20
#> y x xend bin_id seq_id start end file_id strand type feat_id
#> <int> <dbl> <dbl> <chr> <chr> <int> <int> <chr> <chr> <chr> <chr>
#> 1 2 0 473 BVI_008A BVI_0… 1 473 emales… + repe… BVI_00…
#> 2 2 26820 26347 BVI_008A BVI_0… 26348 26820 emales… - repe… BVI_00…
#> 3 3 0 488 BVI_069 BVI_0… 1 488 emales… + repe… BVI_06…
#> 4 3 26808 26320 BVI_069 BVI_0… 26321 26808 emales… - repe… BVI_06…
#> 5 4 0 1081 Cflag_017B Cflag… 1 1081 emales… + repe… Cflag_…
#> 6 4 21311 20230 Cflag_017B Cflag… 20231 21311 emales… - repe… Cflag_…
#> 7 5 0 319 E4-10_086 E4-10… 1 319 emales… + repe… E4-10_…
#> 8 5 20642 20323 E4-10_086 E4-10… 20324 20642 emales… - repe… E4-10_…
#> 9 1 0 458 E4-10_112 E4-10… 1 458 emales… + repe… E4-10_…
#> 10 1 26856 26398 E4-10_112 E4-10… 26399 26856 emales… - repe… E4-10_…
#> 11 6 0 454 RCC970_016B RCC97… 1 454 emales… + repe… RCC970…
#> 12 6 20152 19698 RCC970_016B RCC97… 19699 20152 emales… - repe… RCC970…
#> # ℹ 9 more variables: introns <list>, parent_ids <list>, source <chr>,
#> # score <chr>, phase <int>, name <chr>, width <chr>, geom_id <chr>,
#> # .marginal <lgl>
# get "seqs" track (always track #1)
gg %>% pull_seqs()
#> # A tibble: 6 × 12
#> # Groups: bin_id [6]
#> y x xend strand seq_id bin_id length bin_offset start end file_id
#> <int> <dbl> <dbl> <chr> <chr> <chr> <int> <dbl> <dbl> <int> <chr>
#> 1 6 0 20152 + RCC970_… RCC97… 20152 0 1 20152 emales
#> 2 5 0 20642 + E4-10_0… E4-10… 20642 0 1 20642 emales
#> 3 4 0 21311 + Cflag_0… Cflag… 21311 0 1 21311 emales
#> 4 3 0 26808 + BVI_069 BVI_0… 26808 0 1 26808 emales
#> 5 2 0 26820 + BVI_008A BVI_0… 26820 0 1 26820 emales
#> 6 1 0 26856 + E4-10_1… E4-10… 26856 0 1 26856 emales
#> # ℹ 1 more variable: seq_desc <chr>
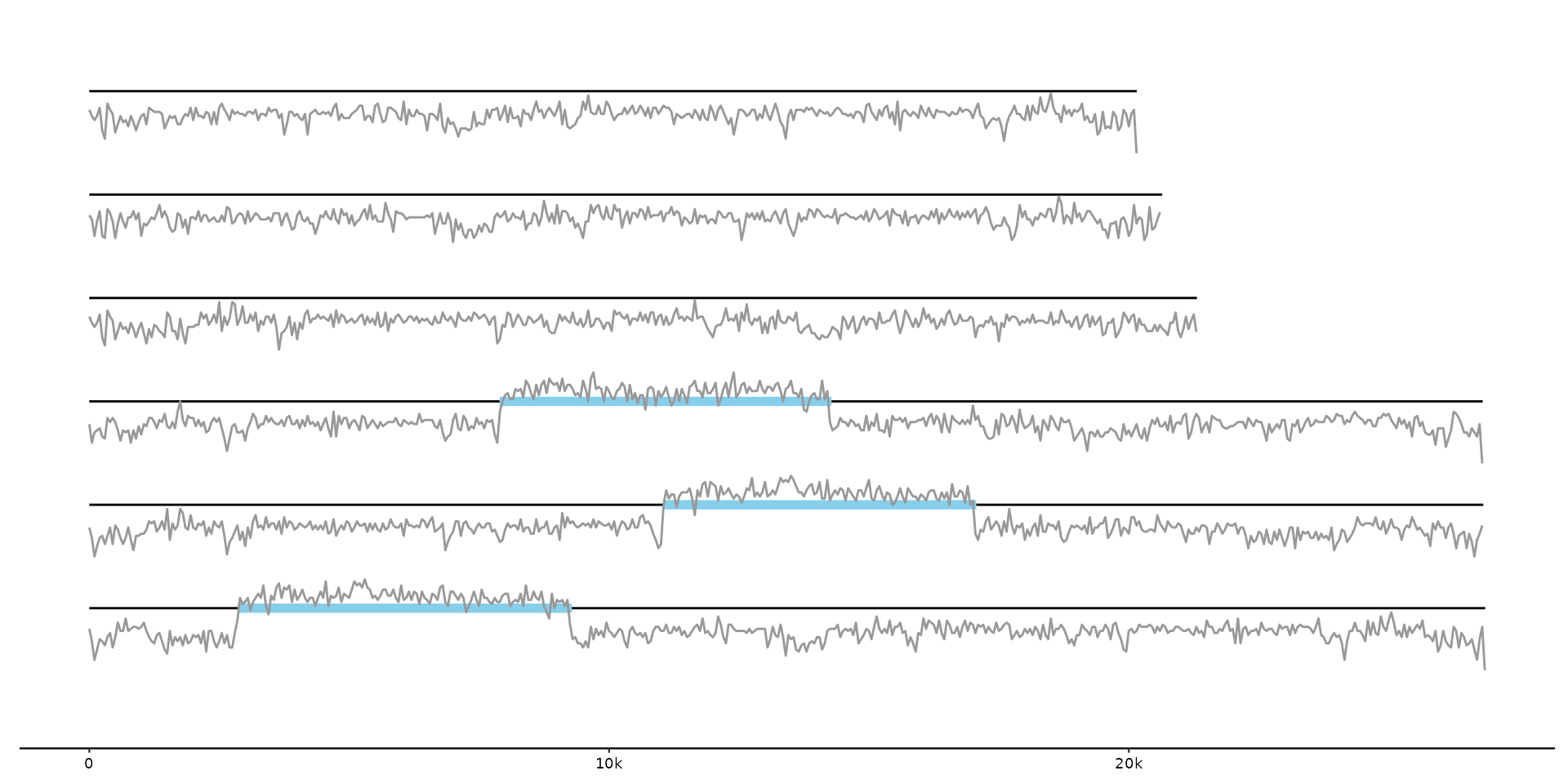
# plot integrated transposons and GC content for some viral genomes
gg <- gggenomes(seqs = emale_seqs, feats = list(emale_ngaros, GC = emale_gc))
gg + geom_seq() +
geom_feat(color = "skyblue") + # defaults to data=feats()
geom_line(aes(x, y + score - .6, group = y), data = feats(GC), color = "gray60")