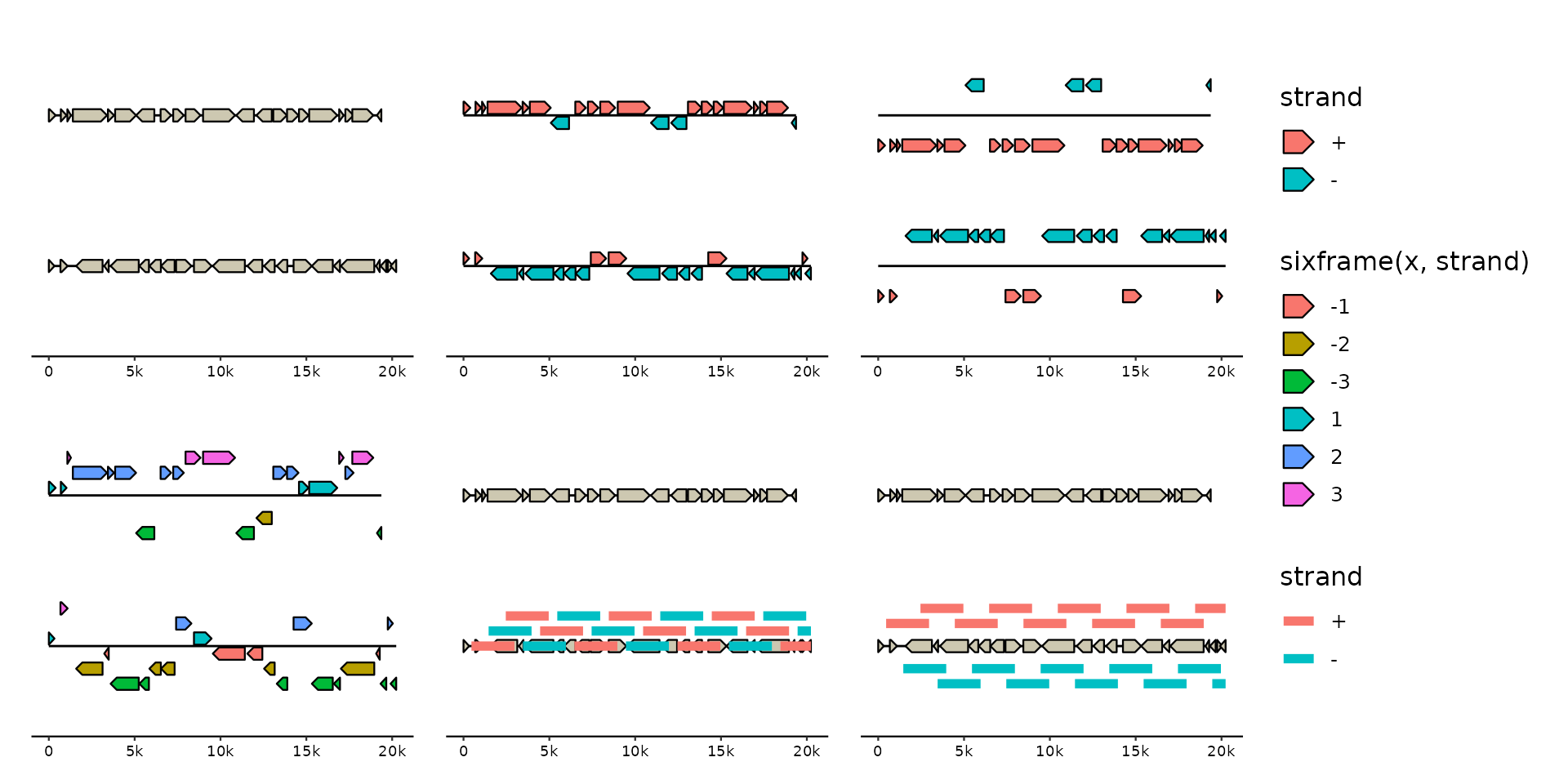
position_strand() offsets forward feats upward and reverse feats downward.
position_pile() stacks overlapping feats upward. position_strandpile()
stacks overlapping feats up-/downward based on their strand.
position_sixframe() offsets the feats based on their strand and reading
frame.
Usage
position_strand(offset = 0.1, flip = FALSE, grouped = NULL, base = offset/2)
position_pile(offset = 0.1, gap = 1, flip = FALSE, grouped = NULL, base = 0)
position_strandpile(
offset = 0.1,
gap = 1,
flip = FALSE,
grouped = NULL,
base = offset * 1.5
)
position_sixframe(offset = 0.1, flip = FALSE, grouped = NULL, base = offset/2)Arguments
- offset
Shift overlapping feats up/down this much on the y-axis. The y-axis distance between two sequences is 1, so this is usually a small fraction, such as 0.1.
- flip
stack downward, and for stranded versions reverse upward.
- grouped
if TRUE feats in the same group are stacked as a single feature. Useful to move CDS and mRNA as one unit. If NULL (default) set to TRUE if data appears to contain gene-ish features.
- base
How to align the stack relative to the sequence. 0 to center the lowest stack level on the sequence, 1 to put forward/reverse sequence one half offset above/below the sequence line.
- gap
If two feats are closer together than this, they will be stacked. Can be negative to allow small overlaps. NA disables stacking.
Value
A ggproto object to be used in geom_gene().
Examples
library(patchwork)
p <- gggenomes(emale_genes) %>%
pick(3:4) + geom_seq()
#> No seqs provided, inferring seqs from feats
f0 <- tibble::tibble(
seq_id = pull_seqs(p)$seq_id[1],
start = 1:20 * 1000,
end = start + 2500,
strand = rep(c("+", "-"), length(start) / 2)
)
sixframe <- function(x, strand) as.character((x %% 3 + 1) * strand_int(strand))
p1 <- p + geom_gene()
p2 <- p + geom_gene(aes(fill = strand), position = "strand")
p3 <- p + geom_gene(aes(fill = strand), position = position_strand(flip = TRUE, base = 0.2))
p4 <- p + geom_gene(aes(fill = sixframe(x, strand)), position = "sixframe")
p5 <- p %>% add_feats(f0) + geom_gene() + geom_feat(aes(color = strand))
p6 <- p %>% add_feats(f0) + geom_gene() + geom_feat(aes(color = strand), position = "strandpile")
p1 + p2 + p3 + p4 + p5 + p6 + plot_layout(ncol = 3, guides = "collect") & ylim(2.5, 0.5)
#> Scale for y is already present.
#> Adding another scale for y, which will replace the existing scale.
#> Scale for y is already present.
#> Adding another scale for y, which will replace the existing scale.
#> Scale for y is already present.
#> Adding another scale for y, which will replace the existing scale.
#> Scale for y is already present.
#> Adding another scale for y, which will replace the existing scale.
#> Scale for y is already present.
#> Adding another scale for y, which will replace the existing scale.
#> Scale for y is already present.
#> Adding another scale for y, which will replace the existing scale.